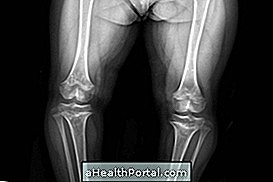
Marote-Lamijev sindrom ali mukopolisaharidoza VI je redka podedovana bolezen, pri kateri imajo nosilci naslednje značilnosti:
- Nizka stopnja,
- obrazne deformacije,
- kratki vrat,
- ponavljajoči se otitis,
- bolezni v dihalnih poteh,
- skeletne malformacije in
- togost mišic.
Bolezen povzročajo spremembe encima Arylsulfatase B, ki preprečujejo, da bi opravljal svojo funkcijo, ki bi razkrila polisaharide, ki se nato kopičijo v celicah in razvijejo simptome, značilne za to boleznijo.
Bolniki s sindromom imajo normalno inteligenco, zato otrok ne potrebuje posebne šole, samo prilagojenih materialov, ki olajšajo interakcijo z učitelji in sošolci.
Diagnozo izdeluje genetik, ki temelji na kliničnem vrednotenju in laboratorijskih biokemičnih analizah. Diagnoza v prvih letih življenja je zelo pomembna za pripravo načrta zgodnjega posredovanja, ki bo pripomogel k razvoju otroka in pri naslavljanju staršev na gensko svetovanje, saj tvegajo, da bo bolezen prenesla na poznejše otroke.
Ni zdravila za Maroteaux-Lamyjev sindrom, vendar se nekatera zdravljenja, kot sta presaditev kostnega mozga in nadomestno zdravljenje z encimi, izkažejo za učinkovite pri zmanjševanju simptomov. Fizikalna terapija se uporablja za zmanjšanje togosti mišic in povečanje telesnih gibov. Vsi bolniki ne prikazujejo vseh simptomov bolezni, resnost se razlikuje od posameznika do posameznika, nekateri pa lahko živijo relativno normalno življenje.